Alzheimer's disease is a neurodegenerative disease whose prevalence is rising. Alzheimer's disease is characterized by the accumulation of beta-amyloid and neurofibrillary tangles, neuronal cholesterol signaling, vascular dysfunction, impaired neurotransmitter activity, and neuroinflammation.
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by a gradual decline in cognitive function, particularly the early involvement of short-term memory.1 Between 1990 and 2019, the incidence and prevalence of Alzheimer's disease (AD) and other forms of dementia increased significantly worldwide by 148% and 161%, respectively. The global mortality rate attributable to Alzheimer's disease and other forms of dementia also goes up from 0.56 million in 1990 to 1.62 million in 2019.2
AD is characterized by an abnormal structure of amyloid beta protein and neurofibrillary tangles in the brain. The deposition of both of these components in the brain causes neuronal atrophy and mortality. The accumulation of the nuclear protein β-amyloid (Aβ42) results in the formation of senile plaques as extracellular lesions of Alzheimer's disease. Neurofibrillary tangles are observed in intraneuronal Alzheimer's disease. 3
The amyloid beta protein originates from the amyloid precursor protein (APP) via enzymatic proteolysis. The gene, known as APP, is located on chromosome 21. The APP overexpression eventually results in increased production and deposition of amyloid peptides (Aβ peptides) in the cerebral cortex. 3 Astrocytes are responsible for the synthesis and transport of cholesterol to neurons, a process facilitated by apolipoprotein E. Reduced neuronal cholesterol levels are expected to inhibit Aβ accumulation, allowing astrocytes to modulate Aβ accumulation via cholesterol signaling.4 Clearance of beta-amyloid requires endothelial cells and pericytes from the blood-brain barrier, intracellular and extracellular enzymatic degradation, brain interstitial fluid (ISF) bulk flow, and the absorption of cerebrospinal fluid (CSF).5
The pathophysiology of Aβ is believed to be the trigger or facilitator for downstream molecular pathways, including tau misfolding, tau-mediated toxicity, tangles accumulation, and tau dissemination, which all result in cortical neuronal degeneration.5 The neurofibrillary tangle consisted of phosphorylated tau protein (P-tau). P-tau themselves are generated from MAPT gene alternative splicing on chromosome 17 (17q21).3
The brain's capillaries, arteries, and arterioles are susceptible to amyloid beta accumulation.3 This accumulation will result in the degeneration of blood vessel wall components and a reduction in blood flow. Cerebral hypoperfusion is reported to be a risk factor for dementia in the general population.6 At the same time, amyloid beta protein also stimulates endothelial cell apoptosis. Vascular endothelial cell growth factor (VEGF) is abundantly expressed in the Central Nervous System. It is found that VEGF substantially inhibited endothelial apoptosis induced by Aβ in vitro.7
The function and survival of cholinergic, serotonergic, noradrenergic, and dopaminergic neurons can be obstructed by Aβ deposits. 3 These disturbances have been demonstrated in numerous previous postmortem investigations. According to those studies AD patients have deficiencies in monoamine neurotransmitters such as noradrenaline, serotonin, and dopamine and decreased acetylcholine transferase.8–10
AD pathophysiology also includes biphasic inflammatory pathway events. As a protective response, microglia phagocytose Aβ fibrils in the early phases, which end as a failure. Following the accumulation of tau tangles, the second neurotoxic stage of microglia is activated.1 Neuroinflammation induced by Aβ peptide deposition stimulates the production of proinflammatory cytokines that induce tau protein phosphorylation.3
This short and up-to-date pathophysiology review shows that genes, chromosomes, neurotransmitters, inflammatory cytokines and chemokines, neuronal lipid metabolism, and brain vascularization may be investigated to determine Alzheimer's disease incidence. More hypotheses, new therapy research, and clinical trials will emerge in the future along with the studies.
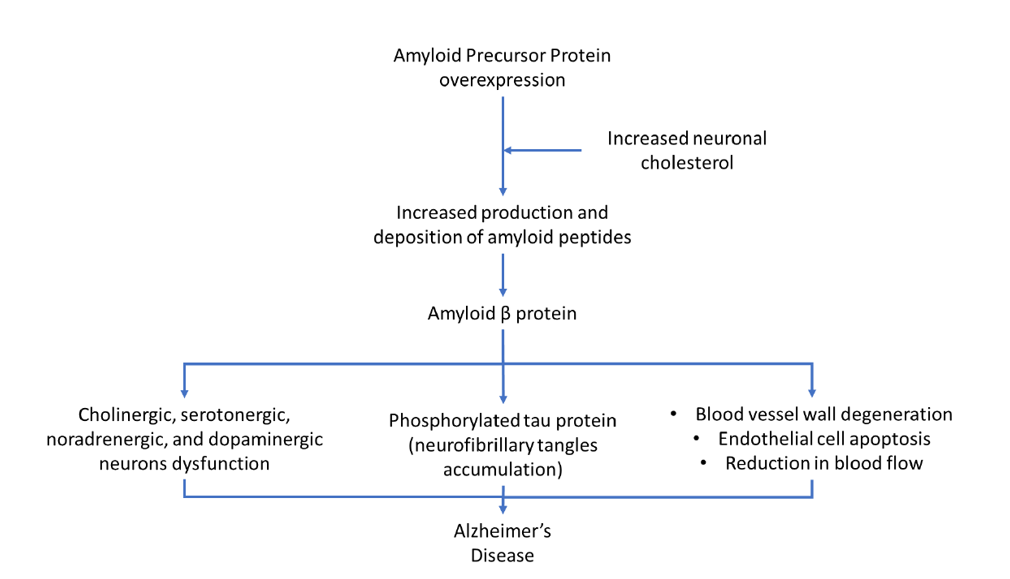
Figure1. Pathophysiology of Alzheimer's Disease (adapted from3, 4, 5, 6, 7, 8, 9, 10)
This article was authored by Valentinus Besin, MD, PhD
Figure1. Pathophysiology of Alzheimer's Disease (adapted from3, 4, 5, 6, 7, 8, 9, 10)
This article was authored by Valentinus Besin, MD, PhD